
Comparative Analysis of PacBio and Oxford Nanopore Sequencing Technologies for Transcriptomic Landscape Identification of Penaeus monodon

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection and RNA Preparation
2.2. PacBio Library Preparation and Sequencing
2.3. ONT Library Preparation and Sequencing
2.4. ONT Self-Correction of Reads
2.5. Transcriptome Analyses: Assembly of High-Quality Long-Reads Using StringTie2
2.6. Correlation Analysis of Gene Expression Level
2.7. Assessment of Final Transcriptomes for Completeness and Functional Assignment
3. Results and Discussion
3.1. Assessment of Characteristics of PacBio SMRT and ONT Read Libraries
3.2. Analysis of Aligned Reads
3.3. Error Correction for ONT Reads
3.4. Transcript Assembly and Identification of New Transcripts Using StringTie2
3.5. Correlation of Gene Expression Levels between ONT and PacBio Samples
3.6. Comparison of Final Isoform Estimation and Functional Annotation
4. Final Remarks
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jenjaroenpun, P.; Wongsurawat, T.; Pereira, R.; Patumcharoenpol, P.; Ussery, D.W.; Nielsen, J.; Nookaew, I. Complete genomic and transcriptional landscape analysis using third-generation sequencing: A case study of Saccharomyces cerevisiae CEN.PK113-7D. Nucleic Acids Res. 2018, 46, e38. [Google Scholar] [CrossRef]
- Bayega, A.; Wang, Y.C.; Oikonomopoulos, S.; Djambazian, H.; Fahiminiya, S.; Ragoussis, J. Transcript Profiling Using Long-Read Sequencing Technologies. In Gene Expression Analysis: Methods and Protocols; Raghavachari, N., Garcia-Reyero, N., Eds.; Springer: New York, NY, USA, 2018; pp. 121–147. [Google Scholar] [CrossRef]
- Giolai, M.; Paajanen, P.; Verweij, W.; Witek, K.; Jones, J.D.G.; Clark, M.D. Comparative analysis of targeted long read sequencing approaches for characterization of a plant’s immune receptor repertoire. BMC Genom. 2017, 18, 564. [Google Scholar] [CrossRef] [PubMed]
- Puglia, G.D.; Prjibelski, A.D.; Vitale, D.; Bushmanova, E.; Schmid, K.J.; Raccuia, S.A. Hybrid transcriptome sequencing approach improved assembly and gene annotation in Cynara cardunculus (L.). BMC Genom. 2020, 21, 317. [Google Scholar] [CrossRef] [PubMed]
- Tilgner, H.; Raha, D.; Habegger, L.; Mohiuddin, M.; Gerstein, M.; Snyder, M. Accurate Identification and Analysis of Human mRNA Isoforms Using Deep Long Read Sequencing. G3 Genes Genomes Genet. 2013, 3, 387–397. [Google Scholar] [CrossRef] [PubMed]
- Amarasinghe, S.L.; Su, S.; Dong, X.; Zappia, L.; Ritchie, M.E.; Gouil, Q. Opportunities and challenges in long-read sequencing data analysis. Genom. Biol. 2020, 21, 30. [Google Scholar] [CrossRef]
- Depledge, D.P.; Srinivas, K.P.; Sadaoka, T.; Bready, D.; Mori, Y.; Placantonakis, D.G.; Mohr, I.; Wilson, C.A. Direct RNA sequencing on nanopore arrays redefines the transcriptional complexity of a viral pathogen. Nat. Commun. 2019, 10, 754. [Google Scholar] [CrossRef]
- Byrne, A.; Beaudin, A.E.; Olsen, H.E.; Jain, M.; Cole, C.; Palmer, T.; DuBois, R.M.; Forsberg, E.C.; Akeson., M.; Vollmers, C. Nanopore long-read RNAseq reveals widespread transcriptional variation among the surface receptors of individual B cells. Nat. Commun. 2017, 8, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Oikonomopoulos, S.; Wang, Y.C.; Djambazian, H.; Badescu, D.; Ragoussis, J. Benchmarking of the Oxford Nanopore MinION sequencing for quantitative and qualitative assessment of cDNA populations. Sci. Rep. 2016, 6, 1–13. [Google Scholar] [CrossRef]
- Seki, M.; Katsumata, E.; Suzuki, A.; Sereewattanawoot, S.; Sakamoto, Y.; Mizushima-Sugano, J.; Sugano, S.; Kohno, T.; Frith, M.C.; Tsuchihara, K.; et al. Evaluation and application of RNA-Seq by MinION. DNA Res. 2019, 26, 55–65. [Google Scholar] [CrossRef]
- Cui, J.; Shen, N.; Lu, Z.; Xu, G.; Wang, Y.; Jin, B. Analysis and comprehensive comparison of PacBio and nanopore-based RNA sequencing of the Arabidopsis transcriptome. Plant Methods 2020, 16, 85. [Google Scholar] [CrossRef]
- Stark, R.; Grzelak, M.; Hadfield, J. RNA sequencing: The teenage years. Nat. Rev. Genet. 2019, 20, 631–656. [Google Scholar] [CrossRef]
- Roach, N.P.; Sadowski, N.; Alessi, A.F.; Timp, W.; Taylor, J.; Kim, J.K. The full-length transcriptome of C. elegans using direct RNA sequencing. Genome Res. 2020, 30, 299–312. [Google Scholar] [CrossRef] [PubMed]
- Wenger, A.M.; Peluso, P.; Rowell, W.J.; Chang, P.-C.; Hall, R.J.; Concepcion, G.T.; Ebler, J.; Fungtammasan, A.; Kolesnikov, A.; Olson, N.D.; et al. Accurate circular consensus long-read sequencing improves variant detection and assembly of a human genome. Nat. Biotechnol. 2019, 37, 1155–1162. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.; Koren, S.; Miga, K.H.; Quick, J.; Rand, A.C.; Sasani, T.A.; Tyson, J.R.; Beggs, A.D.; Dilthey, A.T.; Fiddes, I.T.; et al. Nanopore sequencing and assembly of a human genome with ultra-long reads. Nat. Biotechnol. 2018, 36, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Jain, C.; Aluru, S. A comprehensive evaluation of long read error correction methods. BMC Genom. 2020, 21, 889. [Google Scholar] [CrossRef]
- Pootakham, W.; Uengwetwanit, T.; Sonthirod, C.; Sittikankaew, K.; Karoonuthaisiri, N. A Novel Full-Length Transcriptome Resource for Black Tiger Shrimp (Penaeus monodon) Developed Using Isoform Sequencing (Iso-Seq). Front. Mar. Sci. 2020, 7, 172. [Google Scholar] [CrossRef]
- Yuan, J.; Zhang, X.; Liu, C.; Yu, Y.; Wei, J.; Li, F.; Xiang, J. Genomic resources and comparative analyses of two economical penaeid shrimp species, Marsupenaeus japonicus and Penaeus monodon. Mar. Genom. 2018, 39, 22–25. [Google Scholar] [CrossRef]
- Van Quyen, D.; Gan, H.M.; Lee, Y.P.; Nguyen, D.D.; Nguyen, T.H.; Tran, X.T.; Nguyen, V.S.; Khang, D.D.; Austin, C.M. Improved genomic resources for the black tiger prawn (Penaeus monodon). Mar. Genom. 2020, 100751. [Google Scholar] [CrossRef]
- Uengwetwanit, T.; Pootakham, W.; Nookaew, I.; Sonthirod, C.; Angthong, P.; Sittikankaew, K.; Rungrassamee, W.; Arayamethakorn, S.; Wongsurawat, T.; Jenjaroenpun, P.; et al. A chromosome-level assembly of the black tiger shrimp (Penaeus monodon) genome facilitates the identification of novel growth-associated genes. Mol. Ecol. Resour. 2021, 21, 1620–1640. [Google Scholar] [CrossRef]
- Kovaka, S.; Zimin, A.V.; Pertea, G.M.; Razaghi, R.; Salzberg, S.L.; Pertea, M. Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Gen. Biol. 2019, 20, 278. [Google Scholar] [CrossRef]
- De Coster, W.; D’Hert, S.; Schultz, D.T.; Cruts, M.; Van Broeckhoven, C. NanoPack: Visualizing and processing long-read sequencing data. Bioinformatics 2018, 34, 2666–2669. [Google Scholar] [CrossRef] [PubMed]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Gen. Res. 2017, 27, 722–736. [Google Scholar] [CrossRef] [PubMed]
- Salmela, L.; Walve, R.; Rivals, E.; Ukkonen, E. Accurate self-correction of errors in long reads using de Bruijn graphs. Bioinformatics 2017, 33, 799–806. [Google Scholar] [CrossRef] [PubMed]
- Lima, L.; Marchet, C.; Caboche, S.; Da Silva, C.; Istace, B.; Aury, J.-M.; Touzet, H.; Chikhi, R. Comparative assessment of long-read error correction software applied to Nanopore RNA-sequencing data. Brief Bioinf. 2020, 21, 1164–1181. [Google Scholar] [CrossRef]
- Wyman, D.; Mortazavi, A. TranscriptClean: Variant-aware correction of indels, mismatches and splice junctions in long-read transcripts. Bioinformatics 2019, 35, 340–342. [Google Scholar] [CrossRef]
- Xiao, C.-L.; Chen, Y.; Xie, S.-Q.; Chen, K.-N.; Wang, Y.; Han, Y.; Luo, F.; Xie, Z. MECAT: Fast mapping, error correction, and de novo assembly for single-molecule sequencing reads. Nat. Methods 2017, 14, 1072–1074. [Google Scholar] [CrossRef]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup the Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Quinlan, A.R. BEDTools: The Swiss-Army Tool for Genome Feature Analysis. Curr. Prot. Bioinf. 2014, 47, 11.12.1–11.12.34. [Google Scholar] [CrossRef]
- Okonechnikov, K.; Conesa, A.; García-Alcalde, F. Qualimap 2: Advanced multi-sample quality control for high-throughput sequencing data. Bioinformatics 2016, 32, 292–294. [Google Scholar] [CrossRef]
- Neph, S.; Kuehn, M.S.; Reynolds, A.P.; Haugen, E.; Thurman, R.E.; Johnson, A.K.; Rynes, E.; Maurano, M.T.; Vierstra, J.; Thomas, S.; et al. BEDOPS: High-performance genomic feature operations. Bioinformatics 2012, 28, 1919–1920. [Google Scholar] [CrossRef] [PubMed]
- Pertea, G.; Pertea, M. GFF Utilities: GffRead and GffCompare. F1000Research 2020, 9, 304. [Google Scholar] [CrossRef]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef]
- Tang, A.D.; Soulette, C.M.; van Baren, M.J.; Hart, K.; Hrabeta-Robinson, E.; Wu, C.J.; Brooks, A.N. Full-length transcript characterization of SF3B1 mutation in chronic lymphocytic leukemia reveals downregulation of retained introns. Nat. Commun. 2020, 11, 1438. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing. 2013. Available online: http://www.R-project.org/ (accessed on 1 August 2020).
- Seppey, M.; Manni, M.; Zdobnov, E.M. BUSCO: Assessing Genome Assembly and Annotation Completeness. Methods Mol. Biol. 2019, 1962, 227–245. [Google Scholar] [CrossRef]
- Zdobnov, E.M.; Tegenfeldt, F.; Kuznetsov, D.; Waterhouse, R.M.; Simão, F.A.; Ioannidis, P.; Seppey, M.; Loetscher, A.; Kriventseva, E.V. OrthoDB v9.1: Cataloging evolutionary and functional annotations for animal, fungal, plant, archaeal, bacterial and viral orthologs. Nucl. Acids Res. 2017, 45, D744–D749. [Google Scholar] [CrossRef]
- Moriya, Y.; Itoh, M.; Okuda, S.; Yoshizawa, A.C.; Kanehisa, M. KAAS: An automatic genome annotation and pathway reconstruction server. Nucl. Acids Res. 2007, 35, W182–W185. [Google Scholar] [CrossRef]
- The UniProt Consortium. UniProt: The universal protein knowledgebase in 2021. Nucl. Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef]
- Sessegolo, C.; Cruaud, C.; Da Silva, C.; Cologne, A.; Dubarry, M.; Derrien, T.; Lacroix, V.; Aury, J.M. Transcriptome profiling of mouse samples using nanopore sequencing of cDNA and RNA molecules. Sci. Rep. 2019, 9, 14908. [Google Scholar] [CrossRef]
- Soneson, C.; Yao, Y.; Bratus-Neuenschwander, A.; Patrignani, A.; Robinson, M.D.; Hussain, S. A comprehensive examination of Nanopore native RNA sequencing for characterization of complex transcriptomes. Nat. Commun. 2019, 10, 1–14. [Google Scholar] [CrossRef]
- Rodríguez-Martín, B.; Palumbo, E.; Marco-Sola, S.; Griebel, T.; Ribeca, P.; Alonso, G.; Rastrojo, A.; Aguado, B.; Guigó, R.; Djebali, S. ChimPipe: Accurate detection of fusion genes and transcription-induced chimeras from RNA-seq data. BMC Genom. 2017, 18, 7. [Google Scholar] [CrossRef]
- Weirather, J.L.; de Cesare, M.; Wang, Y.; Piazza, P.; Sebastiano, V.; Wang, X.-J.; Buck, D.; Au, K. Comprehensive comparison of Pacific Biosciences and Oxford Nanopore Technologies and their applications to transcriptome analysis. F1000Research 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Gershman, A.; Romer, T.G.; Fan, Y.; Razaghi, R.; Smith, W.A.; Timp, W. De novo genome assembly of the Tobacco Hornworm moth (Manduca sexta). bioRxiv 2020. [Google Scholar] [CrossRef]
- Zeng, D.; Chen, X.; Peng, J.; Yang, C.; Peng, M.; Zhu, W.; Xie, D.; He, P.; Wei, P.; Lin, Y.; et al. Single-molecule long-read sequencing facilitates shrimp transcriptome research. Sci. Rep. 2018, 8, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Burset, M.; Guigó, R. Evaluation of gene structure prediction programs. Genomics 1996, 34, 353–367. [Google Scholar] [CrossRef]
- Leelatanawit, R.; Sittikankeaw, K.; Yocawibun, P.; Klinbunga, S.; Roytrakul, S.; Aoki, T.; Hirono, I.; Menasveta, P. Identification, characterization and expression of sex-related genes in testes of the giant tiger shrimp Penaeus monodon. Comp. Biochem. Physiol. Part A Mol. Integr. Physiol. 2009, 152, 66–76. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Wei, P.; Zhang, B.; Zhao, Y.; Zeng, D.; Chen, X.; Li, M.; Chen, X. Gonadal transcriptomic analysis and differentially expressed genes in the testis and ovary of the Pacific white shrimp (Litopenaeus vannamei). BMC Genom. 2015, 16, 1006. [Google Scholar] [CrossRef] [PubMed]
- Chandler, J.C.; Aizen, J.; Elizur, A.; Battaglene, S.C.; Ventura, T. Male Sexual Development and the Androgenic Gland: Novel Insights through the de novo Assembled Transcriptome of the Eastern Spiny Lobster, Sagmariasus verreauxi. Sex. Dev. 2015, 9, 338–354. [Google Scholar] [CrossRef] [PubMed]
- Chandler, J.C.; Aizen, J.; Fitzgibbon, Q.P.; Elizur, A.; Ventura, T. Applying the Power of Transcriptomics: Understanding Male Sexual Development in Decapod Crustacea. Integr. Comp. Biol. 2016, 56, 1144–1156. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Hui, M.; Cui, Z.; Luo, D.; Song, C.; Li, Y.; Liu, L. Comparative Transcriptome Analysis Reveals Sex-Biased Gene Expression in Juvenile Chinese Mitten Crab Eriocheir sinensis. PLoS ONE 2015, 10, e0133068. [Google Scholar] [CrossRef]
- Perry, J.C.; Harrison, P.W.; Mank, J.E. The Ontogeny and Evolution of Sex-Biased Gene Expression in Drosophila melanogaster. Mol. Biol. Evol. 2014, 31, 1206–1219. [Google Scholar] [CrossRef]
- Ardui, S.; Ameur, A.; Vermeesch, J.R.; Hestand, M.S. Single molecule real-time (SMRT) sequencing comes of age: Applications and utilities for medical diagnostics. Nucl. Acids Res. 2018, 46, 2159–2168. [Google Scholar] [CrossRef]
- Huerlimann, R.; Wade, N.M.; Gordon, L.; Montenegro, J.D.; Goodall, J.; McWilliam, S.; Tinning, M.; Siemering, K.; Giardina, E.; Donovan, D.; et al. De novo assembly, characterization, functional annotation and expression patterns of the black tiger shrimp (Penaeus monodon) transcriptome. Sci. Rep. 2018, 8, 13553. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Zhang, Y.; Cui, H.; Liu, J.; Wu, Y.; Cheng, Y.; Xu, H.; Huang, X.; Li, S.; Zhou, A.; et al. WEGO 2.0: A web tool for analyzing and plotting GO annotations, 2018 update. Nucl. Acids Res. 2018, 46, W71–W75. [Google Scholar] [CrossRef] [PubMed]
- Jenjaroenpun, P.; Wongsurawat, T.; Wadley, T.D.; Wassenaar, T.M.; Lui, J.; Dai, Q.; Wanchai, V.; Akel, N.S.; Jamshidi-Parsian, A.; Franco, A.T.; et al. Decoding the epitranscriptional landscape from Native RNA sequences. Nucl. Acids Res. 2021, 49, e7. [Google Scholar] [CrossRef]
- Zuo, C.; Blow, M.; Sreedasyam, A.; Kuo, R.C.; Ramamoorthy, G.K.; Torres-Jerez, I.; Li, G.; Wang, M.; Dilworth, D.; Barry, K.; et al. Revealing the transcriptomic complexity of switchgrass by PacBio long-read sequencing. Biotechnol. Biofuels 2018, 11, 170. [Google Scholar] [CrossRef]
- Byrne, A.; Cole, C.; Volden, R.; Vollmers, C. Realizing the potential of full-length transcriptome sequencing. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2019, 374, 20190097. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
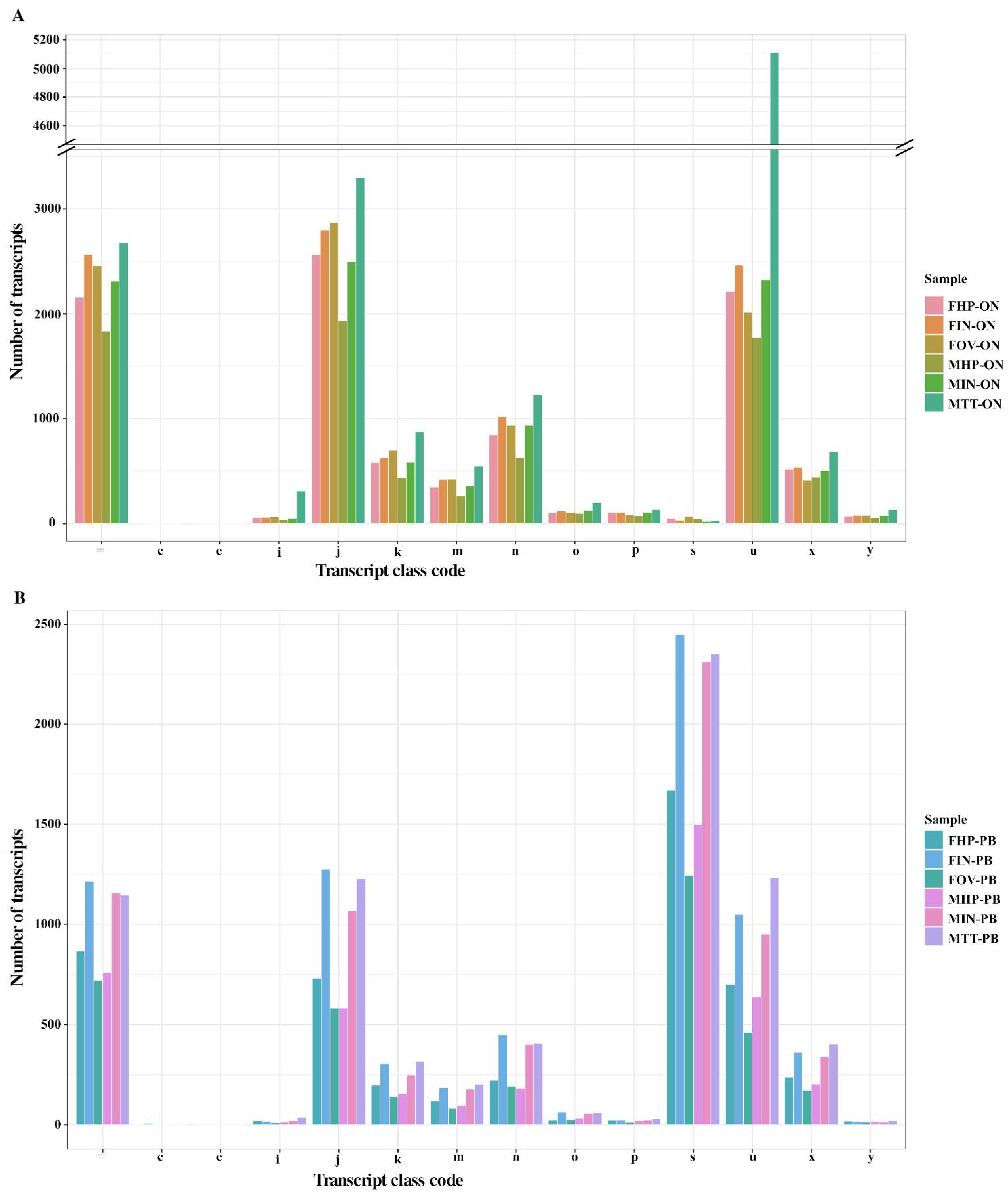{kind=link}
{kind=link}
{kind=link}
| Sample ID | Platform | Sex | Tissue | Reads, Total Bases (Gb) | Read Number | Average Read Length (b) | Read Length N50 |
|---|---|---|---|---|---|---|---|
| FHP_PB | PacBio | Female | Hepatopancreas | 3562 | 1,550,060 | 2297 | 2487 |
| FIN_PB | PacBio | Female | Intestine | 4980 | 1,967,020 | 2531 | 2962 |
| FOV_PB | PacBio | Female | Ovary | 4553 | 1,589,542 | 2864 | 3752 |
| MHP_PB | PacBio | Male | Hepatopancreas | 5199 | 2,701,407 | 1924 | 2367 |
| MIN_PB | PacBio | Male | Intestine | 4221 | 1,949,961 | 2164 | 2738 |
| MTT_PB | PacBio | Male | Testis | 4787 | 2,081,856 | 2299 | 3023 |
| FHP_ON | ONT | Female | Hepatopancreas | 12,465 | 10,556,858 | 1181 | 1681 |
| FIN_ON | ONT | Female | Intestine | 5973 | 5,944,869 | 1005 | 1230 |
| FOV_ON | ONT | Female | Ovary | 14,093 | 8,359,907 | 1686 | 2613 |
| MHP_ON | ONT | Male | Hepatopancreas | 8178 | 6,570,975 | 1245 | 1845 |
| MIN_ON | ONT | Male | Intestine | 5541 | 5,344,621 | 1037 | 1325 |
| MTT_ON | ONT | Male | Testis | 10,919 | 9,223,658 | 1184 | 1746 |
| Sample ID | Read Number | Average of Reads Length | Mapped Bases (%)/Clipped (%) | Mapped Reads (%) | Unmapped Reads | Supplementary Alignments | General Error Rate | |
|---|---|---|---|---|---|---|---|---|
| ONT filtered | FHP_ON | 7,794,835 | 1147 | 99.57/96.51 | 99.48 | 40,170 | 273,464 | 0.12 |
| FIN_ON | 5,237,429 | 996 | 99.01/93.18 | 98.84 | 60,905 | 290,981 | 0.12 | |
| FOV_ON | 5,641,133 | 1719 | 98.73/90.59 | 97.68 | 130,620 | 596,976 | 0.12 | |
| MHP_ON | 4,925,279 | 1222 | 99.61/96.90 | 99.46 | 26,745 | 143,951 | 0.12 | |
| MIN_ON | 3,883,617 | 1029 | 99.25/93.62 | 99.16 | 32,594 | 185,252 | 0.12 | |
| MTT_ON | 6,720,316 | 1185 | 98.78/95.00 | 98.24 | 118,541 | 288,694 | 0.12 | |
| TranscriptClean | FHP_ON | 7,794,835 | 1174 | 99.58/96.59 | 99.48 | 40,170 | 273,464 | 0.02 |
| FIN_ON | 5,237,429 | 1016 | 99.03/93.31 | 98.84 | 60,905 | 275,315 | 0.02 | |
| FOV_ON | 5,641,133 | 1728 | 98.74/90.64 | 97.68 | 130,620 | 596,976 | 0.02 | |
| MHP_ON | 4,925,279 | 1247 | 99.62/96.97 | 99.46 | 26,745 | 143,951 | 0.01 | |
| MIN_ON | 3,883,617 | 1050 | 99.26/93.75 | 99.16 | 32,594 | 185,252 | 0.02 | |
| MTT_ON | 6,720,316 | 1207 | 98.80/95.09 | 98.24 | 118,541 | 288,694 | 0.02 | |
| MECAT | FHP_ON | 712,368 | 1395 | 97.10/92.63 | 96.5 | 24,736 | 114,199 | 0.06 |
| FIN_ON | 715,312 | 1282 | 96.42/91.45 | 95.64 | 31,215 | 125,218 | 0.06 | |
| FOV_ON | 1,701,918 | 1747 | 97.37/92.31 | 96.2 | 64,661 | 242,980 | 0.05 | |
| MHP_ON | 438,541 | 1321 | 96.94/92.41 | 96.06 | 17,286 | 60,668 | 0.06 | |
| MIN_ON | 427,321 | 1327 | 96.29/91.49 | 95.64 | 18,613 | 71,757 | 0.06 | |
| MTT_ON | 1,033,153 | 1346 | 97.34/92.82 | 96.73 | 33,799 | 163,571 | 0.07 |
| Base Level | Intron Chain Level | Transcript Level | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Reference File | Merged File | Reference File | Merged File | Reference File | Merged File | |||||||
| S | P | S | P | S | P | S | P | S | P | S | P | |
| FHP_ON | 100 | 57.7 | 99.9 | 96.7 | 100 | 60.1 | 100 | 99.7 | 99.5 | 59.7 | 100 | 96.8 |
| FIN_ON | 100 | 57.7 | 99.9 | 96.7 | 100 | 60.1 | 100 | 99.7 | 99.5 | 59.6 | 100 | 96.7 |
| FOV_ON | 100 | 57.7 | 99.9 | 96.7 | 100 | 60.1 | 100 | 99.7 | 99.5 | 59.7 | 100 | 96.8 |
| MHP_ON | 100 | 57.7 | 99.9 | 96.7 | 100 | 60.1 | 100 | 99.7 | 99.5 | 59.6 | 100 | 96.7 |
| MIN_ON | 100 | 57.7 | 99.9 | 96.7 | 100 | 60.1 | 100 | 99.7 | 99.5 | 59.7 | 100 | 96.7 |
| MTT_ON | 100 | 57.7 | 99.9 | 96.8 | 100 | 60.1 | 100 | 99.7 | 99.5 | 59.6 | 100 | 96.7 |
| FHP_ONt | 100 | 57.7 | 99.9 | 96.7 | 100 | 60.1 | 100 | 99.7 | 99.5 | 59.7 | 100 | 96.8 |
| FIN_ONt | 100 | 57.7 | 99.9 | 96.7 | 100 | 60.1 | 100 | 99.7 | 99.5 | 59.6 | 100 | 96.7 |
| FOV_ONt | 100 | 57.7 | 99.9 | 96.7 | 100 | 60.1 | 100 | 99.7 | 99.5 | 59.7 | 100 | 96.8 |
| MHP_ONt | 100 | 57.7 | 99.9 | 96.7 | 100 | 60.1 | 100 | 99.7 | 99.5 | 59.6 | 100 | 96.7 |
| MIN_ONt | 100 | 57.7 | 99.9 | 96.7 | 100 | 60.1 | 100 | 99.7 | 99.5 | 59.7 | 100 | 96.7 |
| MTT_ONt | 100 | 57.7 | 99.9 | 96.8 | 100 | 60.1 | 100 | 99.7 | 99.5 | 59.6 | 100 | 96.7 |
| FHP_PB | 100 | 57.7 | 99.9 | 96.8 | 100 | 60.1 | 100 | 99.7 | 99.5 | 59.7 | 100 | 96.8 |
| FIN_PB | 100 | 57.7 | 99.9 | 96.8 | 100 | 60.1 | 100 | 99.7 | 99.5 | 59.7 | 100 | 96.8 |
| FOV_PB | 100 | 57.7 | 99.9 | 96.8 | 100 | 60.1 | 100 | 99.7 | 99.5 | 59.7 | 100 | 96.8 |
| MHP_PB | 100 | 57.7 | 99.9 | 96.8 | 100 | 60.1 | 100 | 99.7 | 99.5 | 59.7 | 100 | 96.8 |
| MIN_PB | 100 | 57.7 | 99.9 | 96.8 | 100 | 60.1 | 100 | 99.7 | 99.5 | 59.7 | 100 | 96.8 |
| MTT_PB | 100 | 57.7 | 99.9 | 96.8 | 100 | 60.1 | 100 | 99.7 | 99.5 | 59.7 | 100 | 96.8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Udaondo, Z.; Sittikankaew, K.; Uengwetwanit, T.; Wongsurawat, T.; Sonthirod, C.; Jenjaroenpun, P.; Pootakham, W.; Karoonuthaisiri, N.; Nookaew, I. Comparative Analysis of PacBio and Oxford Nanopore Sequencing Technologies for Transcriptomic Landscape Identification of Penaeus monodon. Life 2021, 11, 862. https://doi.org/10.3390/life11080862
Udaondo Z, Sittikankaew K, Uengwetwanit T, Wongsurawat T, Sonthirod C, Jenjaroenpun P, Pootakham W, Karoonuthaisiri N, Nookaew I. Comparative Analysis of PacBio and Oxford Nanopore Sequencing Technologies for Transcriptomic Landscape Identification of Penaeus monodon. Life. 2021; 11(8):862. https://doi.org/10.3390/life11080862
Chicago/Turabian StyleUdaondo, Zulema, Kanchana Sittikankaew, Tanaporn Uengwetwanit, Thidathip Wongsurawat, Chutima Sonthirod, Piroon Jenjaroenpun, Wirulda Pootakham, Nitsara Karoonuthaisiri, and Intawat Nookaew. 2021. "Comparative Analysis of PacBio and Oxford Nanopore Sequencing Technologies for Transcriptomic Landscape Identification of Penaeus monodon" Life 11, no. 8: 862. https://doi.org/10.3390/life11080862